Home • Installing • How To Use • Citation
⭐ 2025 Google PhD Fellowship in Health Research awarded to support outstanding and innovative research in computer science and related fields, providing total funding of USD 30.000 over two years - [Link].
⭐ ISME Scholar Mobility Fund awarded with funding of € 2.300 for a research period in July 2026 at the Helmholtz Centre for Environmental Research (UFZ) in Leipzig, Germany
The prediction of biological sequence properties has traditionally relied on alignment-based methods that assume evolutionary homology and depend on curated reference databases. This, in turn, limits scalability and sensitivity for large or heterogeneous datasets, remote homologs, short sequences, and rapidly evolving genomic regions. Although Machine-Learning (ML) approaches offer alignment-free alternatives, their broader adoption is limited by: (i) the lack of standardized, externally validated benchmark models across diverse datasets, and (ii) the technical expertise required for feature engineering, model selection, and evaluation. Automated machine learning (AutoML) alleviates these challenges by systematically optimizing representations and models with minimal user intervention. However, most existing frameworks prioritize task-specific model construction and lack mechanisms for preserving trained models as persistent, comparable benchmarks. We introduce BioAutoML-FAST, an end-to-end web platform for automated ML analysis of nucleotide and amino acid sequences. It supports both classification and regression tasks and automates feature extraction, model training, and evaluation without requiring prior user expertise. Uniquely, it serves as a community benchmarking resource, hosting a continuously expanding repository of reusable, standardized models (currently 60) for genomic, transcriptomic, and proteomic applications. Extensive validation on independent datasets demonstrates performance comparable to or exceeding that of state-of-the-art methods, including protein language models such as ESM-2. BioAutoML-FAST is available at https://bioautoml.icmc.usp.br/. This website is free and open to all users, and there is no login requirement.
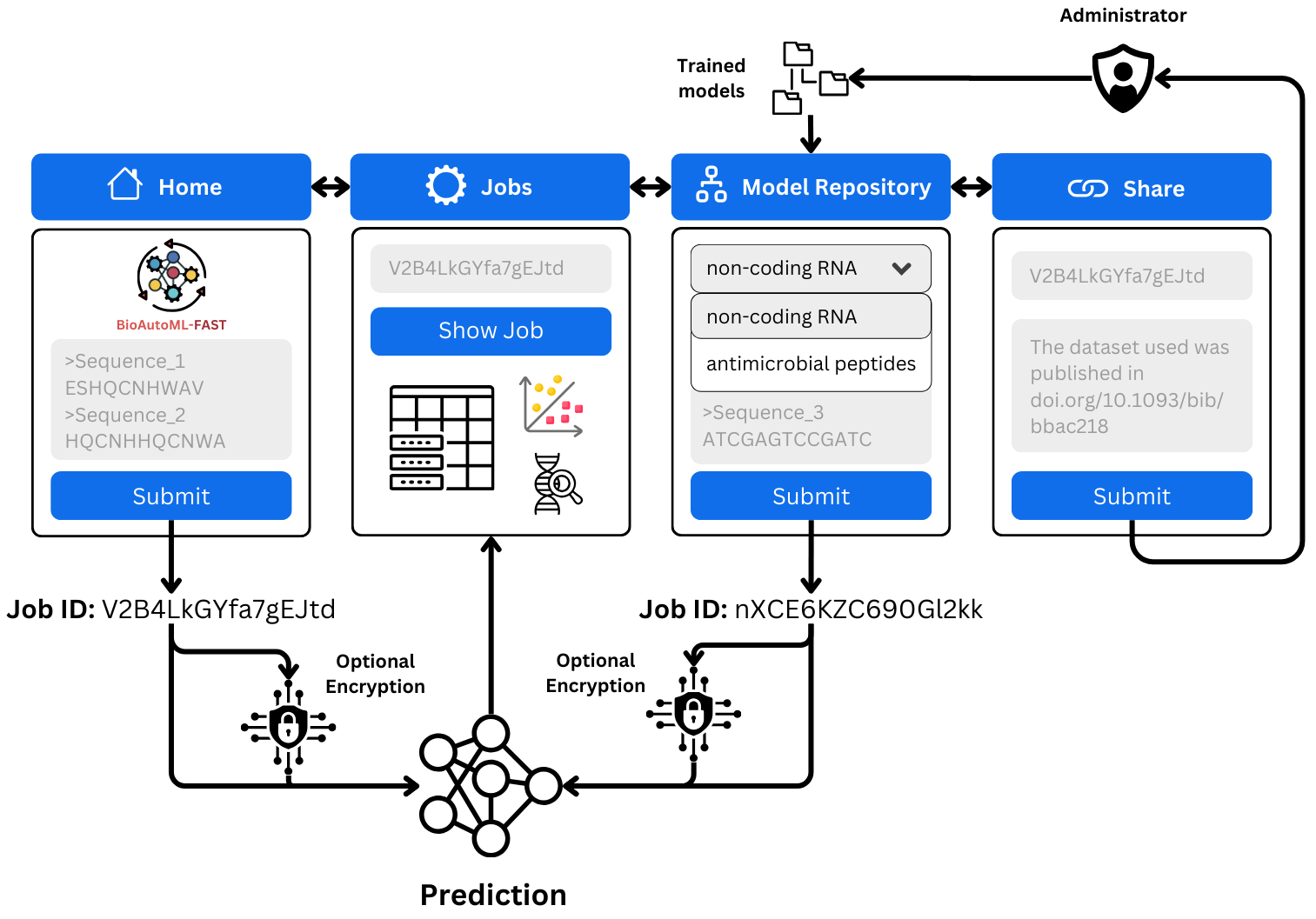
-
Breno L. S. de Almeida, Robson P. Bonidia, Martin Bole, Anderson P. Avila-Santos, Peter F. Stadler, Ulisses Rocha, André C. P. L. F. de Carvalho
-
Correspondence: brenoslivio@usp.br, bonidia@utfpr.edu.br or ulisses.rocha@ufz.de
Silva de Almeida, B. L., Bonidia, R., Bole, M., Avila-Santos, A., Stadler, P. F., Nunes da Rocha, U., & de Carvalho, A. C. L. F. (2026). BioAutoML-FAST: an automated machine-learning platform for reusable and benchmarked biological sequence models. bioRxiv, 2026-04. DOI
If you want to use BioAutoML-FAST locally you can clone the repository and add the necessary submodules:
git clone https://github.com/Bonidia/BioAutoML-FAST.git BioAutoML-FAST
cd BioAutoML-FAST
git submodule init
git submodule update1 - Install uv
If using Linux or Mac:
curl -LsSf https://astral.sh/uv/install.sh | shIf using Windows, use irm to download the script and execute it with iex:
powershell -ExecutionPolicy ByPass -c "irm https://astral.sh/uv/install.ps1 | iex"2 - Preparing the virtual environment
With uv installed, inside the folder use following command to syncronize the virtual environment with the necessary dependencies:
uv sync3 - Activate environment
After preparing the environment, you can activate the environment on Linux or Mac with:
source .venv/bin/activateUsing Windows:
.venv\Scripts\activate4 - Deactivate environment
You can deactivate the environment using:
deactivateThere are two main scripts that are part of BioAutoML-FAST, engineering.py and generation.py. engineering.py is the first step of BioAutoML-FAST, with optimal descriptors selection, and generation.py is the second step, with hyperparamer optimization:
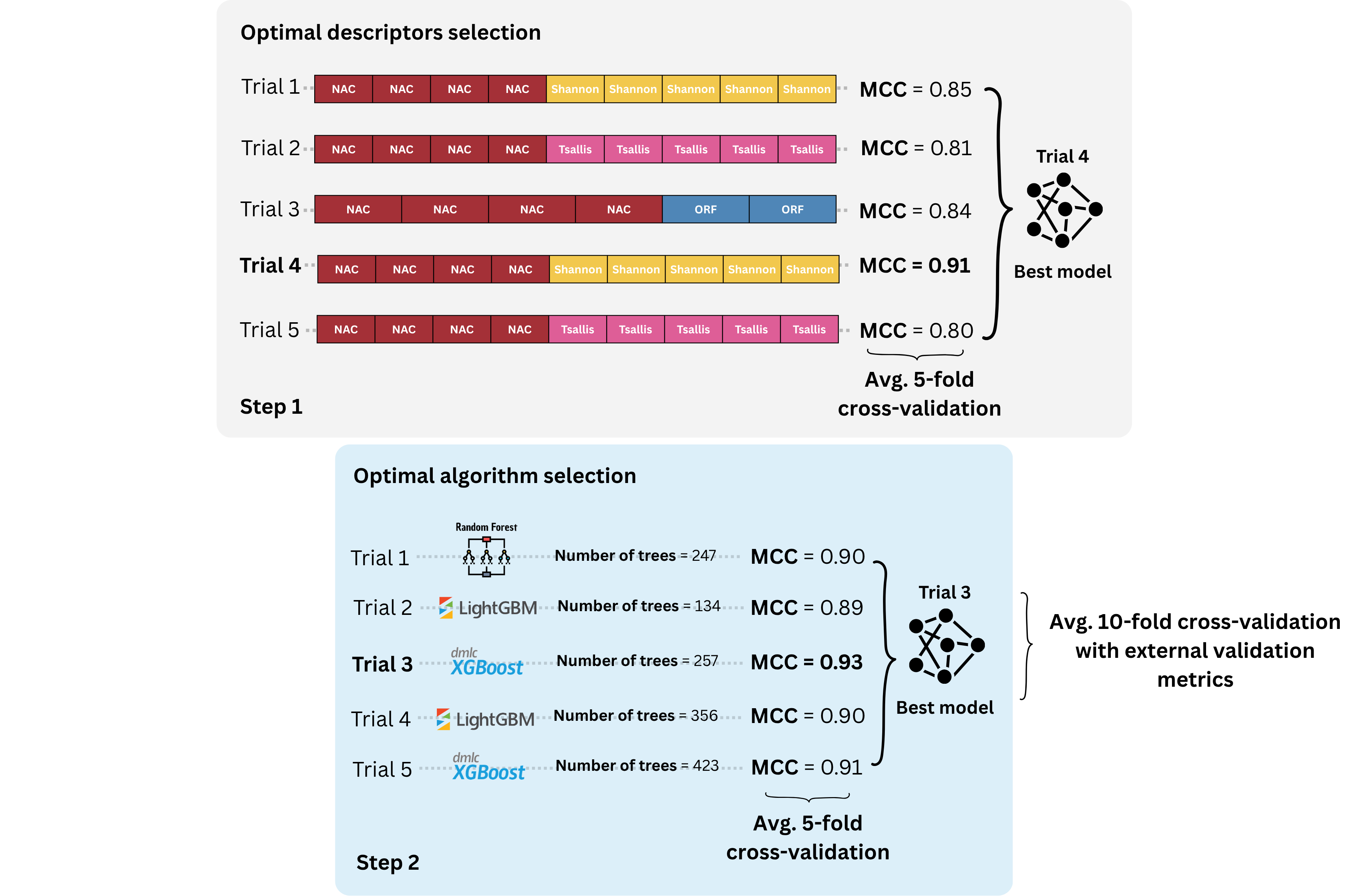
The engineering.py script performs the first step of BioAutoML-FAST. It extracts sequence descriptors from the input FASTA files, performs automated feature engineering/descriptor selection, and then automatically calls generation.py for model generation and hyperparameter optimization.
| Option | Description | Default |
|---|---|---|
-fasta_train, --fasta_train |
One or more training FASTA files. | Required |
-fasta_label_train, --fasta_label_train |
Labels associated with each training FASTA file. The order must match -fasta_train. |
Required |
-fasta_test, --fasta_test |
One or more testing FASTA files. | Optional |
-fasta_label_test, --fasta_label_test |
Labels associated with each testing FASTA file. The order must match -fasta_test. |
Optional |
-dtype, --dtype |
Type of input data. Supported values: DNA/RNA or Protein. |
DNA/RNA |
-task, --task |
Machine learning task. Use 0 for classification and 1 for regression. |
0 |
-estimations, --estimations |
Number of estimations used during automated feature engineering. | 200 |
-patience, --patience |
Number of trials without improvement before early stopping. | 80 |
-tuning, --tuning |
Number of trials used for hyperparameter optimization in generation.py. |
150 |
-difference, --difference |
Minimum improvement required before early stopping. | 0.001 |
-n_cpu, --n_cpu |
Number of CPU cores to use. Use -1 to use all available cores. |
-1 |
-output, --output |
Output directory where results will be saved. | Required |
python engineering.py \
-fasta_train train/ncRNA.fasta train/lncRNA.fasta train/circRNA.fasta \
-fasta_label_train ncRNA lncRNA circRNA \
-fasta_test test/ncRNA.fasta test/lncRNA.fasta test/circRNA.fasta \
-fasta_label_test ncRNA lncRNA circRNA \
-dtype DNA/RNA \
-task 0 \
-output resultspython engineering.py \
-fasta_train train/enzyme.fasta \
-fasta_label_train enzyme \
-fasta_test test/enzyme.fasta \
-fasta_label_test enzyme \
-dtype Protein \
-task 1 \
-output resultsThe generation.py script performs the second step of BioAutoML-FAST. It trains and optimizes machine learning models using the descriptors generated during the feature engineering step. The module supports both classification and regression tasks, including hyperparameter optimization and external test evaluation.
| Option | Description | Default |
|---|---|---|
-path_model, --path_model |
Path to a previously trained model to be reused for prediction or evaluation. | '' |
-task, --task |
Machine learning task. Use 0 for classification and 1 for regression. |
0 |
-tuning, --tuning |
Number of hyperparameter optimization trials. | 150 |
-train, --train |
Training feature matrix in CSV format. | Required |
-train_label, --train_label |
Training labels in CSV format. | Required |
-train_nameseq, --train_nameseq |
CSV file containing sequence names/identifiers for the training set. | Required |
-test, --test |
Test feature matrix in CSV format. | Optional |
-test_label, --test_label |
Test labels in CSV format. | Optional |
-test_nameseq, --test_nameseq |
CSV file containing sequence names/identifiers for the test set. | Optional |
-n_cpu, --n_cpu |
Number of CPU cores to use. Use -1 to use all available cores. |
-1 |
-output, --output |
Output directory where models and results will be saved. | Required |
Note: This script can be used directly with structured data, without the need of the first step.
If you use this code in a scientific publication, we would appreciate citations to the following paper:
Silva de Almeida, B. L., Bonidia, R., Bole, M., Avila-Santos, A., Stadler, P. F., Nunes da Rocha, U., & de Carvalho, A. C. L. F. (2026). BioAutoML-FAST: an automated machine-learning platform for reusable and benchmarked biological sequence models. bioRxiv, 2026-04. DOI
@article{silva2026bioautoml,
title={BioAutoML-FAST: an automated machine-learning platform for reusable and benchmarked biological sequence models},
author={Silva de Almeida, Breno Livio and Bonidia, Robson and Bole, Martin and Avila-Santos, Anderson and Stadler, Peter F and Nunes da Rocha, Ulisses and de Carvalho, Andre CP L F},
journal={bioRxiv},
pages={2026--04},
year={2026},
publisher={Cold Spring Harbor Laboratory}
}